未来计划
更多关于 Rosetta@home 的详情,请看这里。
介绍
我们现在研究工作的目标是开发一种分子内和分子间的改良模型,并用这个模型来预测和设计大分子结构及其交互作用。这种预测和设计的应用,不仅自身就具有相当重要的生物学意义,而且为改进模型加深基础性理解提供了严格且客观的测试。
我们使用名为 Rosetta 的计算机程序来给出蛋白质及其预测的计算。在 Rosetta
的核心内,有若干功能和方法。它们包括:计算大分子内或大分子间交互作用所产生能量的势函数,发现氨基酸序列或蛋白质联合体最低能量结构的方法(即蛋白质结构预测),及发现蛋白质或蛋白质联合体所需最弱能量氨基酸序列的方法(即蛋白质设计)。预测和设计测试的反馈被不断用于改进潜在功能和查找算法。开发一种能处理这些不同问题的计算机程序具有相当大的好处。首先,这些不同的应用提供了基础物理模型的补充性测试(当然,基础物理/物理化学也是一样的)。其次,许多热门问题,如弹性脊椎蛋白质设计和具有脊椎弹性的蛋白质嵌合,都涉及了对不同最优化方法的结合。
蛋白质结构设计
在过去的数年里,我们利用我们的计算性蛋白质设计方法,重新设计了每一个序列的残基,从而戏剧般地使一些小蛋白质稳定下来,还重新设计了蛋白质骨架构造,将单节显性蛋白质转化成二聚交换捆,掌握了对酶的热稳定方法。重点是对蛋白质 G 的折叠路径的重新设计。蛋白质 G 是一个包含了被单 alpha 螺旋分隔的双 beta 折叠的小蛋白质。在自然形成的蛋白质中,第一个折叠被分裂,而第二个折叠在限制速度里生成。在重新设计的变量中,第一个折叠是极为稳定的,第二个却是不稳定的,其顺序完全相反:第一个折叠形成时,第二个折叠在转换状态里被分裂。蛋白质折叠路径的重新设计能力客观地说明了我们对蛋白质决定性因素的了解已经大有进步。
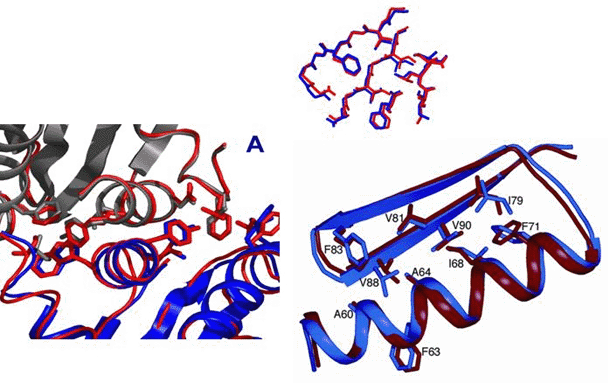
Figure 1: Design of proteins and protein-protein interactions with high-resolution accuracy. Comparison of design model and crystal structure of (left) interface of novel designed endonuclease with new DNA cleavage specificity, and (right) the de novo designed protein TOP7.
Left panel, Tanja Kortemme. Right panel, Gautam Dantas.
|
最近极为令人激动的成果是随机选择三维结构创造的新蛋白质。我们开发了一种通用的计算策略,用于创造这些能将完整的骨架弹性整合进最优化螺旋异构体序列的蛋白质。这些是通过结合 Rosetta 的快速预测过程、精细预测修正和序列设计方法从而完成的。此程序用于设计一个有 93 残基的蛋白质,它被称为 TOP7 ,有全新的序列和拓扑。TOP7被发现是单节显性且折叠的,它的 X 光晶体结构居然和设计模型很相似
(RMSD = 1.2 Å,见图 1 的右侧)。新球型蛋白质折叠设计同设计模型晶体结构的紧密联系扩大了蛋白质设计和蛋白质结构预测之间的关联,打开了探索蛋白质宇宙中未知领域的大门。
Design of Protein-Protein Interactions
To extend these methods to protein-protein interactions and
particularly to the redesign of interaction specificity, we chose the
high-affinity complex between colicin E7 DNase and its cognate
inhibitory immunity protein as a model system. We used the physical
model described above and a modification of our rotamer search-based
computational design strategy to generate novel DNase-inhibitor protein
pairs predicted to interact tightly with one another but not with the
wild-type proteins. The designed protein complexes have subnanomolar
affinities, are functional and specific in vivo, and have more than an
order of magnitude affinity difference between cognate and noncognate
pairs in vitro. This approach should be applicable to the design of
interacting protein pairs with novel specificities for delineating and
reengineering protein interaction networks in living cells.
In collaboration with the research groups of Barry Stoddard and Ray
Monnat (Fred Hutchinson Cancer Research Center),
we generated an artificial, highly specific endonuclease by fusing domains of homing
endonucleases I-DmoI and I-CreI through computational optimization of a
new domain-domain interface between these normally noninteracting
proteins. The resulting enzyme, E-DreI (Engineered I-DmoI/I-CreI),
binds a long chimeric DNA target site with nanomolar affinity, cleaving
it precisely at a rate equivalent to its natural parents. We are
currently trying to generate new endonucleases by extending our design
methodology to protein–nucleic acid interfaces to redesign the
protein-DNA interface.
In both of these systems it has been possible to determine x-ray
crystal structures of the designed complexes. As in the TOP7 case, the
actual structures are very close to the design models (Figure 1, left
panel), which validates the accuracy of our approach to high-resolution
modeling.
Prediction of Protein Structure
The picture of protein folding that motivates our approach to ab
initio protein tertiary structure prediction is that sequence-dependent
local interactions bias segments of the chain to sample distinct sets
of local structures, and that nonlocal interactions select the lowest
free-energy tertiary structures from the many conformations compatible
with these local biases. In implementing the strategy suggested by this
picture, we use different models to treat the local and nonlocal
interactions. Rather than attempting a physical model for local
sequence-structure relationships, we turn to the protein database and
take the distribution of local structures adopted by short sequence
segments (fewer than 10 residues in length) in known three-dimensional
structures as an approximation to the distribution of structures
sampled by isolated peptides with the corresponding sequences.
The primary nonlocal interactions considered are hydrophobic burial,
electrostatics, main-chain hydrogen bonding, and excluded volume.
Structures that are simultaneously consistent with both the local
sequence structure biases and the nonlocal interactions are generated
by using simulated annealing to minimize the nonlocal interaction
energy in the space defined by the local structure distributions.
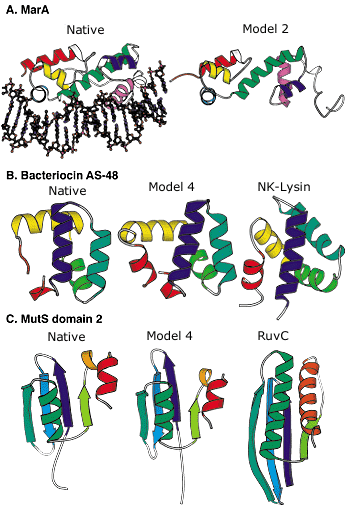
Figure 2: Blind protein structure predictions from CASP3 and CASP4.
A: Left, crystal structure of the MarA transcription factor bound to DNA; right, our best submitted model in CASP3. Despite many incorrect details, the overall fold is predicted with sufficient accuracy to allow insights into the mode of DNA binding.
B: Left, the crystal structure of bacteriocin AS-48; middle, our best submitted model in CASP4; right, a structurally and functionally related protein (NK-lysin) identified using this model in a structure-based search of the Protein Data Bank (PDB). The structural and functional similarity is not recognizable using sequence comparison methods (the identity between the two sequences is only 5 percent).
C: Left, crystal structure of the second domain of MutS; middle, our best submitted model for this domain in CASP4; right, a structurally related protein (RuvC) with a related function recognized using the model in a structure-based search of the PDB. The similarity was not recognized using sequence comparison or fold recognition methods.
Image: Rich Bonneau
|
Rosetta has been tested in the biannual CASP (critical assessment of
structure prediction) experiments in which predictors are challenged to
make blind predictions of the structures adopted by protein sequences
whose structures have been determined but not yet published. Since
CASP3 in 1998, Rosetta has consistently been the top performing method
for ab initio prediction, as reported by independent assessors. In the
CASP4 experiment, for example, Rosetta was tested on 21 proteins. The
predictions for these proteins, which lack detectable sequence
similarity to any protein with a previously determined structure, were
of unprecedented accuracy and consistency. (Some examples are shown in
Figure 2.) Excellent predictions were also made in the CASP5 and CASP6
experiments. Encouraged by these promising results, we generated models
for all large protein families fewer than 150 amino acids in
length.
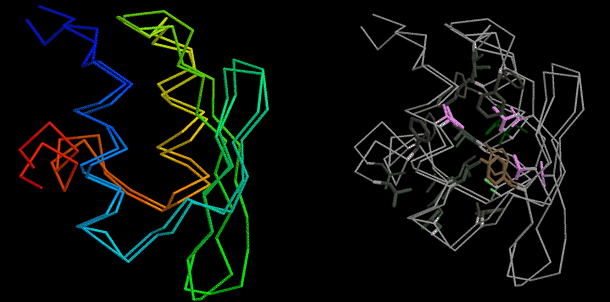
Figure 3: The first close to atomic-level resolution, blind ab initio structure prediction—CASP6 T281. The high-resolution refinement methodology described in the text produced a model 1.5 Å RMSD from the crystal structure (left panel), with aspects of the native side-chain packing (right panel).
Image: Phil Bradley
|
A highlight of CASP6 was the first de novo blind prediction that
used our high-resolution refinement methodology to achieve close to
high-resolution accuracy. The relatively short sequence (76 residues)
allowed us to apply our all-atom refinement methodology not only to the
native sequence but also to the sequence of many homologs. The center
of the lowest energy cluster of structures turned out to be remarkably
close to the native structure (1.5 Å; Figure 3). The-high
resolution refinement protocol decreased the RMSD from 2.2 Å to
1.5 Å, and the side chains pack in a somewhat native-like manner
in the protein core (Figure 3, right panel).
We have extended the Rosetta ab initio structure prediction strategy
to the problem of using limited experimental data to generate models of
proteins. By incorporating chemical shift and NOE information and more
recently dipolar coupling information into the Rosetta structure
generation procedure, we have been able to generate much more accurate
models than with ab initio structure prediction alone or when using the
same limited data sets with conventional nuclear magnetic resonance
(NMR) structure generation methodology. An exciting recent development
is that the Rosetta procedure can also take advantage of unassigned NMR
data and hence circumvent the difficult and tedious step of assigning
NMR spectra.
The Rosetta ab initio structure prediction method, the Rosetta-based
NMR structure determination method, and a new method for comparative
modeling that uses the Rosetta de novo approach to model the parts of a
structure (primarily long loops) that cannot be modeled accurately
based on a homologous structure template have all been implemented in a
public server called Robetta.
This server, which has a constant backlog
of users worldwide, was one of the best all-around fully automated
structure prediction servers in the CASP5 and CASP6 tests.
Prediction of Protein-Protein Interactions
For a number of years we have worked on protein structure refinement,
a challenging problem because of the large number of degrees of
freedom. We became interested in protein-protein docking because, with
the approximation that the two partners do not undergo significant
conformational changes during docking, the space to be
searched—the six rigid-body degrees of freedom in addition to the
side-chain degrees of freedom—is much smaller. While important in
its own right, this problem is a good stepping stone to the harder
structure refinement problem.
We developed a new method to predict protein-protein complexes from
the coordinates of the unbound monomer components. This method employs
a low-resolution, rigid-body, Monte Carlo search followed by
simultaneous optimization of backbone displacement and side-chain
conformations with the Monte Carlo minimization procedure and physical
model used in our high-resolution structure prediction work. The
simultaneous optimization of side-chain and rigid-body degrees of
freedom contrasts with most other current approaches, which model
protein-protein docking as a rigid-body shape-matching problem, with
the side chains kept fixed. We have recently improved the method
(RosettaDock) by developing an algorithm that allows efficient sampling
of off-rotamer side-chain conformations during docking.
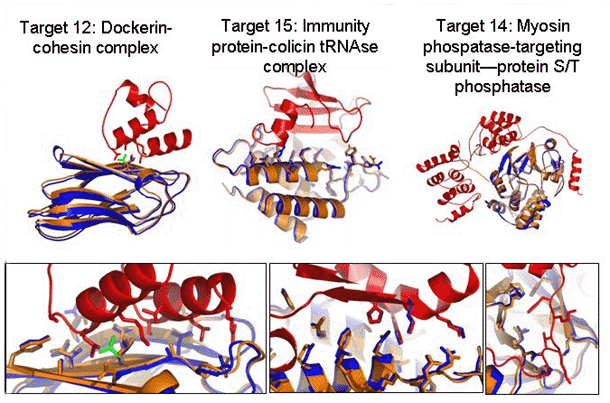
Figure 4: CAPRI (critical assessment of predicted interactions) protein-protein docking results. Superposition of predicted (blue) and x-ray (red and orange) protein complex structures. Green, a side chain whose conformation was correctly predicted to change upon complex formation. Upper panel, whole complex. Lower panel, details of the interface. In addition to the rigid-body orientation, the conformations of most of the side chains are predicted correctly.
Image: Ora Furman
|
The power of RosettaDock was highlighted in the recent blind CAPRI
protein-protein docking challenge that was held in December 2004. In
CAPRI, predictors are given the structures of two proteins known to
form a complex, and challenged to predict the structure of the complex.
RosettaDock predictions for targets without significant backbone
conformational changes were striking, as shown in Figure 4. Not only
were the rigid-body orientations of the two partners predicted nearly
perfectly but also almost all the interface side chains were modeled
very accurately. These correct models clearly stood out as lower in
energy than all other models we generated, which suggests the potential
function is reasonably accurate.
These promising results suggest that the method may soon be useful
for generating models of biologically important complexes from the
structures of the isolated components, and more generally suggest that
high-resolution modeling of structures and interactions is within
reach. A clear goal for our monomeric structure prediction work is to
approach the level of accuracy of these models.
Improvement of Physical Model
Our current approach to improving energy functions involves a
combination of quantum chemistry calculations on simple model
compounds, traditional molecular mechanics approaches, and protein
structural analysis. We have used such an approach to develop an
improved hydrogen-bonding potential. A particularly notable result is
that the orientation dependence of the hydrogen bond in quantum
chemistry calculations on formamide dimers is remarkably similar to
that seen in side-chain–side-chain hydrogen bonds in protein
structures but different from that in current molecular mechanics force
fields, which neglect the covalent character of the hydrogen bond.
Feedback from the prediction and design calculations has provided
continual impetus and guidance for improving the energy function; for
example, inadequacies in our treatment of protein-protein interactions
have led to the recent development of a rotamer-based model for
water-mediated hydrogen bonds.
Plans for the Future
Our prediction and design methods have now reached the point where
they can be applied to important biological problems. Particularly
encouraging after years of work on high-resolution modeling are the
close to atomic resolution predictions of the structures of complexes
in CAPRI (Figure 4), the 1.5-Å de novo prediction in CASP6
(Figure 3), and the close agreement of the TOP7 (Figure 1, right) and
protein-protein interface design models (Figure 1, left) with the x-ray
crystal structures. These results suggest that high-resolution modeling
is starting to work.
In the next several years, we aim to improve and extend our methods.
We are particularly focused on improving the accuracy of
high-resolution structure prediction (which will be required if the
models are to be generally useful). To accomplish this, we will work to
improve the underlying physical model and the sampling methodology. We
are also developing improved methods to predict and redesign
protein-DNA interaction specificity, and extending our protein design
methodology to the design of enzymes that catalyze chemical reactions
not catalyzed by naturally occurring proteins.
Please visit our web site at http://www.bakerlab.org
for additional information including a list of our research publications.